头痛伴发热,别只想到中枢神经系统感染!
原创 作者:仝书严 公号:丁香园神经时间 发布时间:2023-12-15 19:58 发表于北京
原文地址:头痛伴发热,别只想到中枢神经系统感染!
1 病情简介
患者女性,25 岁,因「头痛伴发热半月余」入院。
患者于 2023 年 4 月 19 日受凉后出现发热,体温最高 40℃,伴有头痛,为胀痛,不伴有恶心、呕吐,不伴有咳嗽、咳痰及流涕等症状,在当地医院就诊,考虑普通上感,予以抗感染、退热治疗,患者体温可降低至正常,药效减退后发热及头痛症状反复。
2023 年 4 月 26 日患者开始出现双上肢不自主轻微抖动及视物模糊。
2023 年 5 月 1 日患者出现排尿困难以及排便困难,当地医院给患者完善颅脑、胸部、全腹 CT 平扫:未见明显异常,当地医院予以抗感染及退热药物应用患者症状无明显缓解。
患者为求进一步诊治遂于 2023 年 5 月 5 日至我院急诊就诊,拟诊「中枢神经系统感染?」收入我科。
既往史,个人史,家族史无特殊。
查体:内科系统:体温 38.5℃,脉搏 106 次/min,血压 125/85 mmHg,呼吸 20 次/min,律齐,两肺呼吸音清,未闻及干湿性啰音,下腹部稍膨隆;神经系统:神清,精神差,查体欠配合,双侧瞳孔等大等圆,直径约 3 mm,对光反射灵敏,双眼外展受限,双侧鼻唇沟对称,伸舌居中,四肢肌张力减退,双上肢肌力 5- 级,双下肢肌力 3 级,双侧指鼻试验、跟膝胫试验及 Romberg 征均不能合作,双侧深浅感觉对称存在,双侧上肢腱反射存在,双下肢腱反射消失,双侧 Hoffmann 征阴性,双侧 Babinski 征阴性,颈抵抗,颏胸距 3 指,Kernig 征(+)。
入院完善辅助检查:头颅 MRI:未示异常;脑电图示:慢波增多。
化验:脑脊液压力:250 mmH2O,透明清亮;脑脊液常规:白细胞总数 111 × 10^6/L [正常值 (0~10)× 10^6/L],单个核细胞比例 98.2%,潘氏反应:弱阳性;脑脊液生化:蛋白 1.13 g/L,氯 112.7 mmol/L(正常值 120~132 mmol/L),葡萄糖 2.94 mmol/L(正常值 2.22~3.89 mmol/L)。同步血生化:葡萄糖:8.47 mmol/L(正常值 3.89~6.1 mmol/L),氯 99.9 mmol/L(正常值 96~108 mmol/L);脑脊液细胞学:呈淋巴-单核细胞反应。风湿免疫、肝炎艾滋梅毒、肿瘤指标等相关检验均未示异常。
2 诊疗思路
定位诊断:患者头痛,脑膜刺激征阳性定位于脑膜;患者双眼外展受限,定位于外展神经;患者尿便障碍,双下肢肌力减退,双下肢腱反射消失,定位于腰髓;该患者双上肢肌力肌张力减退,但是腱反射正常,无病理征,考虑发热所致治疗后观察。
定性诊断:感染?自身免疫性?
初步诊治方案:
该患者初步诊断「中枢神经系统感染?」,治疗上暂时先予以阿昔洛韦 500 mg q8h 抗病毒,美罗培南 1 g q8h 抗感染,脱水降颅压等治疗;同时予以完善脑脊液高通量测序(Next-generation sequencing,NGS)及自身免疫性脑炎相关抗体检查。
3 后续检查、结果回报
1)进一步完善腰椎 MRI 增强如下图:
![[]](https://medblog.cn/wp-content/uploads/2023/12/f0f65c46-3fa7-4ae8-b1fa-bd97c8efa53b.png) 图 脊髓 MRI-T1WI 增强:A 图(矢状位)示 T12~L1 脊髓表面线样强化;B 及 C 图(横断面)
图 脊髓 MRI-T1WI 增强:A 图(矢状位)示 T12~L1 脊髓表面线样强化;B 及 C 图(横断面)
该患者在热退后双上肢肌力恢复至 5 级,腱反射正常,无病理征,且患者双下肢对称性弛缓性瘫痪,故无证据支持颈髓相关体征,且考虑患者家庭经济困难,故未完善颈髓平扫及增强检查。
2)脑脊液 NGS 未示有意义结果(检出单纯疱疹病毒 I 型,序列数 1)。
3)自身免疫性脑炎抗体检测结果:血清 :抗胶质纤维酸性蛋白(GFAP)抗体基于细胞底物的实验(CBA)阳性(+)1:32;基于组织底物的实验(TBA)检测(+);脑脊液:TBA 检测,脑脊液(+)。
诊断:自身免疫性胶质纤维酸性蛋白星形胶质细胞病
患者在热退后双上肢肌力恢复至 5 级,予以以上治疗方案后仅有头痛症状有所缓解,于 2023 年 5 月 10 日予以丙种球蛋白(0.4 g/kg)20 g/qd 5 天;甲泼尼龙 500 mg/qd(每 5 天减量),同时补钾补钙,抑酸护胃治疗。该患者于 2023 年 05 月 26 日出院,继续口服激素治疗。
出院情况:双眼球活动自如,双下肢肌力 5 级,排尿困难(导尿状态)及排便困难尚无明显缓解;2023 年 07 月 20 日序贯予以利妥昔单抗应用,激素继续减量,患者排尿困难及排便困难症状改善;2023 年 9 月 5 日随访患者无头痛、无恶心、无排便及排尿困难症状,双眼活动灵活,四肢肌力 5 级,复查腰椎 MRI 未见脊膜强化。
4 讨 论
自身免疫性胶质纤维酸性蛋白星形细胞病(autoimmune glial fibrillary acidic protein astrocytopathy,GFAP-A)是一种以胶质纤维酸性蛋白(glial fibrillary acidic protein,GFAP)抗体为特异性标志物的自身免疫炎性中枢神经系统疾病,由梅奥诊所首次报道并证实,可累及脑、脑膜、脊髓和视神经 [1, 2],临床上常以头痛、发热、脑炎、脊髓炎、视力异常等为主要表现,也可表现为震颤、痴呆、自主神经功能障碍等,常对类固醇激素治疗敏感 [3]。
GFAP 是星形胶质细胞的中间丝蛋白,具有维持星形胶质细胞形态稳定,参与血脑屏障形成,调节突触功能等多种生物学作用 [4, 5]。作为一种细胞内抗原,针对 GFAP 的抗体难以与之相互作用,故学者们认为 GFAP-IgG 很可能自身并无致病力,但其可作为细胞毒性 T 细胞介导的自身免疫反应的标志物,脑脊 GFAP-IgG 具有高度特异度和敏感度,其阳性率和滴度明显高于血清 [6]。
GFAP-A 患者的血清和脑脊液中常重叠其他自身免疫疾病相关的抗体如抗 N-甲基-D-天冬氨酸受体抗体 IgG(NMDAR-IgG)最为常见,其次为抗水通道蛋白 4 抗体 IgG(AQP4-IgG),抗髓鞘少突胶质细胞糖蛋抗体 IgG(MOG-IgG)等 [2, 7, 8]。
GFAP-A 患者 40%~66% 有前驱感染病史,有文献报道该病可继发于疱疹病毒感染之后 [9]。约 20% 的患者可伴有自身免疫性疾病,25% 的患者可伴发肿瘤,以卵巢畸胎瘤最为常见(约 75%),其他癌症罕见且多样,肿瘤可在神经系统症状起病时存在或在之后(多在 2 年内)被发现 [6, 9, 10]。
诊断标准 [6]:
1)急性或亚急性起病,临床表现为脑膜、脑、脊髓、视神经受累或各种症状的组合;2)MRI 可见脑室旁线样放射状强化和(或)脊髓长节段受累伴中央强化病灶;3)脑脊液 GFAP 抗体阳性(CBA 或 TBA);4)脑活体组织检查提示小血管周围炎症伴小胶质细胞活化;5)类固醇激素治疗有效;6)排除其他可能疾病。
鉴别诊断:
1)中枢神经系统感染:GFAP-A 早期最容易被误诊为中枢神经系统感染(包括病毒性脑膜脑炎、结核性脑膜脑炎或细菌性脑膜炎等感染性疾病),往往采取抗感染治疗,有的患者因为存在其他抗体,使得诊断更加模糊。为此,有必要重视疾病之间的鉴别诊断 [11]。
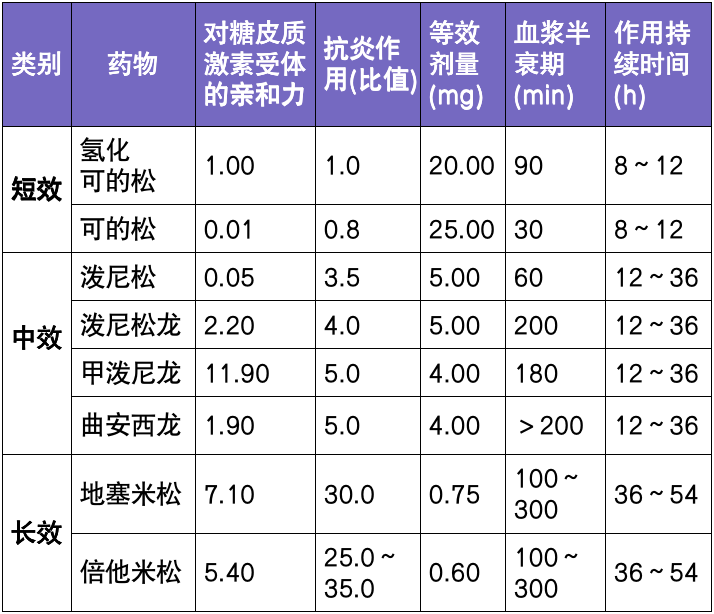
GFAP-A 应与视神经脊髓炎(neuromyelitis optic spectrum disorder, NMOSD)抗髓鞘少突胶质细胞糖蛋白免疫球蛋白 G 抗体相关疾病(antimyelin oligdendrocyte glycoproteinIgG associated disorders,MOGAD),三者同时属于自身免疫细胞病,具体鉴别点如下:
GFAP-A 与 NMOSD 和 MOGAD 的鉴别诊断 [6, 11~18]
![[]](https://medblog.cn/wp-content/uploads/2023/12/e0632ed0-4c79-4bbf-8808-da231b5f2653.jpg)
2)原发性中枢神经系统血管炎(primary angiitis of the central nervous system,PACNS),类固醇激素反应性慢性淋巴细胞性炎症伴脑桥血管周围强化症以及急性播散性脑脊髓炎(acute disseminated encephalomyelitis, ADEM)等疾病进行鉴别诊断 [11]。
治疗及预后:
关于 GFAP-A 的治疗目前尚无循证医学证据,急性期一线免疫治疗推荐大剂量糖皮质激素冲击治疗,急性期治疗包括大剂量类固醇激素冲击治疗、静脉注射免疫球蛋白和血浆置换 [6, 11, 14],有学者提出口服泼尼松从 60 mg/d 或 1 mg/kg/d(不超过 100 mg/d)开始,维持 3 个月,随后用量每月减 10 mg/d 至 10 mg/d,再每月减 1 mg/d 至停用 [6]。
Yang 等人认为长期口服低剂量的类固醇(如甲泼尼龙 4~8 mg/d)是必要的,但仍需进一步研究及验证 [14]。早期减量或停用类固醇激素可能会出现症状和影像学病灶的复发,此时可能需要再次冲击治疗。
对于难治或复发病例可考虑加用免疫抑制剂,如吗替麦考酚酯或者硫唑嘌呤,利妥昔单抗和环磷酰胺也可作为选择。大部分患者预后较好 [10],少数患者对治疗的反应差甚至死亡,一些患者可遗留不同程度的功能残疾 [6, 11]。
总结:临床工作中,神经科医师遇到表现为不明原因的脑膜脑炎、脑炎、脊髓炎、视神经炎或者自主神经功能障碍的患者时,均应尽快进行脑脊液 GFAP 抗体检测,如确诊该病应尽早开始类固醇治疗,以改善症状及预后,同时建议对患者进行至少为期 2 年的肿瘤随访。
本文作者:仝书严 南京医科大学第四临床学院在读博士
指导老师:龚爱平主任医师 许静主任医师 陈浪副主任医师 徐州医科大学第二附属医院
审核专家:陈为安 温州医科大学附属第一医院神经内科 主任医师
更多精彩内容欢迎关注「丁香园神经时间」
每天 20:00 更新
🌟 记得设置星标关注**呦👇
![[]](https://medblog.cn/wp-content/uploads/2023/12/5d53774d-a242-4143-bd98-47e7b148971f.png)
策划 | 时间胶囊
投稿 | zhangjing3@dxy.cn
题图|站酷海洛
参考文献:
1. Fang, B., et al., Autoimmune Glial Fibrillary Acidic Protein Astrocytopathy: A Novel Meningoencephalomyelitis. JAMA Neurol, 2016. 73(11): p. 1297-1307.
2. Flanagan, E.P., et al., Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: Analysis of 102 patients. Ann Neurol, 2017. 81(2): p. 298-309.
3. 梁奇明, et al., 自身免疫性胶质纤维酸性蛋白星形细胞病的临床特征分析. 华中科技大学学报(医学版), 2021. 50(5): p. 620-625.
4. 叶锦龙, et al., 以中枢系统炎症为主要表现的 14 例自身免疫性胶质纤维酸性蛋白星形细胞病患者的临床特征分析. 中风与神经疾病杂志, 2020. 37(12): p. 1101-1104.
5. 郑秀君, et al., 自身免疫性胶质纤维酸性蛋白星形胶质细胞病临床特征分析. 罕见病研究, 2022. 1(2): p. 137-141.
6. 章殷希, et al., 自身免疫性胶质纤维酸性蛋白星形胶质细胞病. 中华神经科杂志, 2020. 53(4): p. 317-320.
7. Shan, F., Y. Long, and W. Qiu, Autoimmune Glial Fibrillary Acidic Protein Astrocytopathy: A Review of the Literature. Front Immunol, 2018. 9: p. 2802.
8. Kimura, A., et al., Clinical characteristics of autoimmune GFAP astrocytopathy. J Neuroimmunol, 2019. 332: p. 91-98.
9. Li, J., et al., Autoimmune GFAP astrocytopathy after viral encephalitis: A case report. Mult Scler Relat Disord, 2018. 21: p. 84-87.
10. Dang, J., S. Lei, and J. Chen, Autoimmune glial fibrillary acidic protein astrocytopathy presented as isolated area postrema symdrome: a case report. BMC Neurol, 2022. 22(1): p. 271.
11. 戚晓昆 and 刁东卫, 自身免疫性胶质纤维酸性蛋白星形胶质细胞病. 中华神经科杂志, 2023. 56(1): p. 82-87.
12. Kunchok, A., A. Zekeridou, and A. McKeon, Autoimmune glial fibrillary acidic protein astrocytopathy. Curr Opin Neurol, 2019. 32(3): p. 452-458.
13. 姚海燕, et al., 自身免疫性胶质纤维酸性蛋白星形细胞病的脑膜脊膜病变研究. 广东医学, 2021. 42(10): p.1203-1206.
14. Yang, X., et al., Treatment of Autoimmune Glial Fibrillary Acidic Protein Astrocytopathy: Follow-Up in 7 Cases. Neuroimmunomodulation, 2017. 24(2): p.113-119.
15. 邱伟, 罗文静, and 周一凡, 重新认识髓鞘少突胶质细胞糖蛋白抗体相关疾病. 中华医学杂志,2020. 100(5): p.3.
16. 管阳太 and 俞昊君, 髓鞘少突胶质细胞糖蛋白抗体相关疾病. 中华神经科杂志, 2022. 55(6): p.7.
17. 中国免疫学会神经免疫分会, 邱伟, and 徐雁, 抗髓鞘少突胶质细胞糖蛋白免疫球蛋白 G 抗体相关疾病诊断和治疗中国专家共识. 中国神经免疫学和神经病学杂志 2020 年 27 卷 2 期 86-95 页 ISTIC PKU CA, 2020.
18.《神经病学》,第 8 版,贾建平,人民卫生出版社.




暂无评论内容